

Many human cancers do not respond to treatment, and often times those that initially respond eventually acquire drug resistance. Our lab uses high-throughput screening technology in combination with tractable pre-clinical mouse models to investigate basic mechanisms of intrinsic and acquired drug resistance. We also use genetic and computational tools to understand the how cancer therapies exert their effects and how to best combine drugs to achieve greater efficacy and preempt the evolution of chemoresistance. Our goal is to identify novel cancer drug targets, as well as strategies for tailoring drug regimens to target protective mechanisms used by cancers to evade and escape cancer therapy.
Microenvironmental Drug Resistance
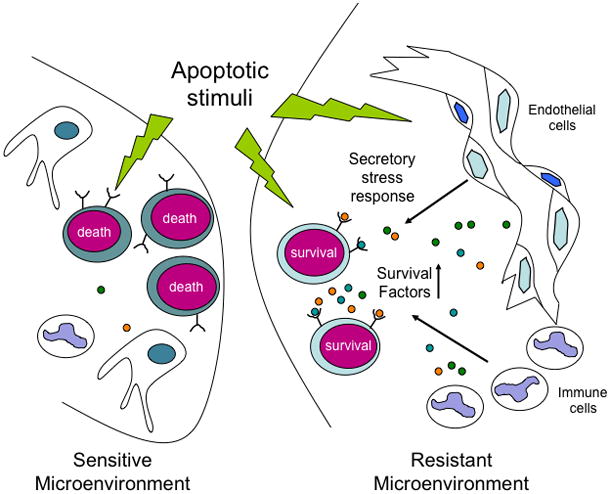
We have demonstrated that factors secreted from the tumor microenvironment can either protect tumor cells from chemotherapy or potentiate chemotherapeutic efficacy. For example, cytokines are released in select tumor-bearing sites in response to DNA damaging agents, creating “chemo-resistant niches” that can promote the persistence of a minimal residual tumor burden and serve as a reservoir for eventual tumor relapse. This kind of cytokine release represents a previously uncharacterized secretory response that also occurs in response to physiological stresses like tissue hypoxia. Thus, conventional chemotherapies can induce tumor regression while simultaneously eliciting stress responses that protect subsets of tumor cells in distinct anatomical locations from drug action. These studies provided the first example of how a cancer can co-opt pro-survival signals that typically support the survival and regeneration of stem and progenitor cells. This process has subsequently been shown to occur in the context of numerous malignancies, including solid tumors. Thus, the processes of cancer cell survival and tissue regeneration are intimately linked in the aftermath of cancer therapy.
In vivo screening for modulators of drug response
We have adapted screening approaches traditionally reserved for single-cell organisms or simple metazoans for use in mammals. Specifically we used loss of function pool-based screening to, for the first time, carry out high-throughput forward genetics in mice in vivo. In some contexts, as many as 25,000 cells contribute to a given malignancy following tail vein injection, and, consequently, nearly 25,000 loss of function phenotypes can be screened in the context of an individual mouse. Notably, the shRNAs or gRNAs that enrich and deplete in vivo differ strikingly from those that enrich and deplete in culture, implicating the tumor microenvironment as a central contributor to tumors homeostasis. These data suggest that large-scale in vivo screens are readily feasible with a limited cohort of mice – an approach that we extensively use in ongoing experiments. Importantly, this strategy can be used to screen both hematopoietic and solid tumors and has led to the identification of critical drug targets and processes in glioblastoma and pancreatic cancers.
Examining single and combination mechanisms of drug action
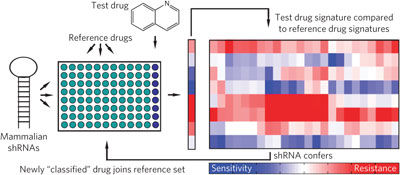
We have developed an RNA interference (RNAi)-based approach to characterize chemotherapeutic function in mammalian cells. Simply put, we could use RNAi-induced phenotypic “signatures” to characterize drug action or the effects of distinct tumor microenvironments on drug action with unprecedented resolution. These robust RNAi-based “signatures” could be used to cluster unknown drugs into functional categories and define mechanisms of action for uncharacterized cytotoxic agents. We have subsequently used this signature approach for applications that were previously not feasible using other drug characterization metrics. For example, we have been able to examine the consequences of adding multiple drugs together in a way that allows us to identify combination mechanisms of actions. This, in turn, has enabled us to start to make predictions as to which drug combinations will be synergistic. We have also used these signatures to examine drug mechanism of action during the course of drug derivatization – the process of improving drug characteristics. Here, we can identify when changes in drug efficacy arise from altered drug pharmacokinetics and pharmacokinetics or, alternatively, changes in drug mechanism. Finally, extensions of this technology have provided a basis for studying tumor heterogeneity and the emergence of resistant clones from heterogeneous tumor populations following therapy.